
Herramientas bioinformáticas para el análisis de secuencias
Published:
Las bases de datos están llenas de secuencias biológicas, pero ¿cómo analizar esa información? Aquí aprenderemos algunos análisis básicos que nos pueden dan información muy útil sobre la Biología.
Introducción
Ya conocimos diferentes bases de datos con información biológica; sin embargo, estos datos solo son útiles si son analizados. Debido a la gran cantidad de datos existentes y a la necesidad de hacer múltiples cálculos y procedimientos rápidamente, la mayoría de análisis de datos biológicos en la actualidad se realizan con la ayuda de programas computacionales y algoritmos capaces de procesas grandes cantidades de información. A pesar de esto, sigue siendo esencial que los investigadores y en general usuarios de estos programas, comprendan aspectos básicos de su funcionamiento y las teorías biológicas en las que se fundamentan para poder interpretar adecuadamente sus resultados.
En este práctica conoceremos algunas de las herramientas más útiles para el análisis de secuencias biológicas.
Objetivos
- Conocer métodos de análisis de secuencias biológicas.
- Conocer herramientas para el análisis de secuencias biológicas.
- Comprender en qué consiste un alineamiento de secuencias y su relevancia para entender la biología de un organismo y su evolución.
Alineamiento de secuencias
El alineamiento de secuencias consiste en hacer concordar lo mejor posible dos o más secuencias, tanto de ADN como de proteína y son una de las formas más comunes de comparar dos o más secuencias biológicas.
¿Cuál es el propósito o la utilidad de hacer un alineamiento de secuencias?
Los alineamientos permiten obtener información filogenética, estructural y funcional, y siguen siendo uno de los principales análisis bioinformáticos realizados en diversos estudios biológicos.
Alineamiento local vs. alineamiento global.
Veamos varios tipos de alineamientos:

¿Cómo alinear secuencias?
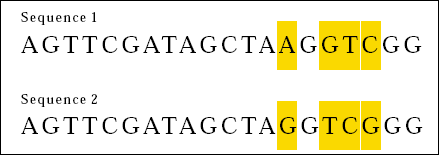
Y si las alineamos así…

Busquemos secuencias (por ejemplo del gen Smaug) en la base de datos OrthoDB y hagamos un alineamiento con MAFFT.
Para visualizar el alineamiento vamos a usar el software gratuito JalView.
BLAST (Basic Local Alignment Search Tool)
BLAST es una herramienta del NCBI que permite hacer búsqueda de secuencias por similaridad, tanto para secuencias de ADN como para secuencias de proteínas. Local significa que se alinean solo segmentos de las secuencias.
Hagamos un BLAST:
>Secuencia misteriosa 1
GCCTCTCATACTAGGAGCTCCTGACATAGCCTTCCCACGATTAAATAATATAAGATTTTGACTACTGCCC
CCCTCTTTAACTCTTCTTATTATAAGAACACTAGCTGATAAAGGAGCAGGAACTGGGTGAACAGTCTACC
CACCTCTGTCTGCAAATTTGGCCCATGAAGGAACATCTGTAGATTTAGCCATCTTCAGTCTCCATATAGC
AGGAGTCTCTTCTATCTTAGGAGCTATAAATTTCATCTCTACAATTATCAATATACGGCCGAAAGGTATA
AGGACAGATCGAATACCTTTATTTGTATGAGCAGTGCTAATCACTGCCATTCTATTACTTCTTTCTTTAC
>Secuencia misteriosa 2
QIHRQISSTSPANRVSPASILASPSPPAPTSPSSSSISVRKKLPSGTKQKPLPPKSSSSKLSSPVAVQDE
IEIEIAEVLYGMMRMPSTSKQEAAGNDLTEAAKSTVEVKSRVSSPISNPQTLPQSSITLAANSSSSNVSA
IAPKRKKPRHVKYEDDNSSRVTTIKSEAEAPSKSQVPFSNQLKSSGSGEGNSSVLDSIIPLTRESNASLD
SEKKENNLSKDETILPKVESSSGFRSDGEGAKSSSPEKEKFEIDLMAPPPVRSSSERGGEMMECVAAEAK
PKVTEVETEAKPLLKEDRSDPAIHDSQEKKRPRMVAEAEHHKFERNCELKLDLDKSDHVGLVNKHHVQKP
PPQQQLSVPDKTAQASHLPLHMSMPGWPGGLPTMGYMAPTQGVVP
>Secuencia misteriosa 3
ACCAGAAGAGCACCGAACTGCTCATCCGCAAGTTGCCCTTCCAGCGACTGGTCCGCGAAATCGCCCAGGA
CTTCAAGACCGACCTACGCTTCCAGAGCTCCGCCGTCATGGCGCTGCAGGAAGCCAGCGAGGCCTACCTG
GTCGGCCTCTTCGAGGATACCAATCTCTGCGCCATCCACGCCAAGCGG
>Secuencia misteriosa 4
TMYLILGAWSAMLGTALSMLIRAELGQPGSLIGDDQIYNVIVTAHAFIMIFFMVMPIMIGGFGNWLVPLM
LGAPDMAFPRLNNMSFWLLPPSLTLLLAGSAVENGAGTGWTVYPPLASNMAHAGASVDLTIFSLHLAGAS
SILGAINFITTVINMRTQGMTMERMPLFVWAVFITAFLLLLSLPVLAGAITMLLTDRNLNTSFFDPAGGG
DPILYQHLFWFFGHP
>Secuencia misteriosa 5
ILYFMFGMWAGMIGMSMSLIIRMELSTSGSILKNDQIYNGMVTLHAFIMIFFMVMPIMIGGFGNWLIPLM
LGAPDMAFPRMNNMSFWLLIPSLLFLLMSGIINTGVGTGWTMYPPLSSLIGHNSISIDMSIFSLHLAGAS
SIMGAINFISTIFNMN
Para una información más detallada sobre cómo usar BLAST descarga este documento.
Búsqueda de homología con HMMER
En muchas ocasiones nos interesa conocer la estructura o función de una secuencia de ADN o de una proteína. Un paso importante para conocer mejor las secuencias de nuestro interés es compararla con otras secuencias conocidas y detectar homologías (es decir, parentesco o relaciones evolutivas). HMMER es un programa que usa modelos estadísticos basados en cadenas de Markov para comparar una secuencia de interés con secuencias depositadas en las bases de datos para detectar homologías.
Online vs. Local
¿Es mejor usar herramientas disponibles en la web o instalar el programa en nuestro propio computador?

Recursos adicionales
https://viralzone.expasy.org/e_learning/alignments/content.html